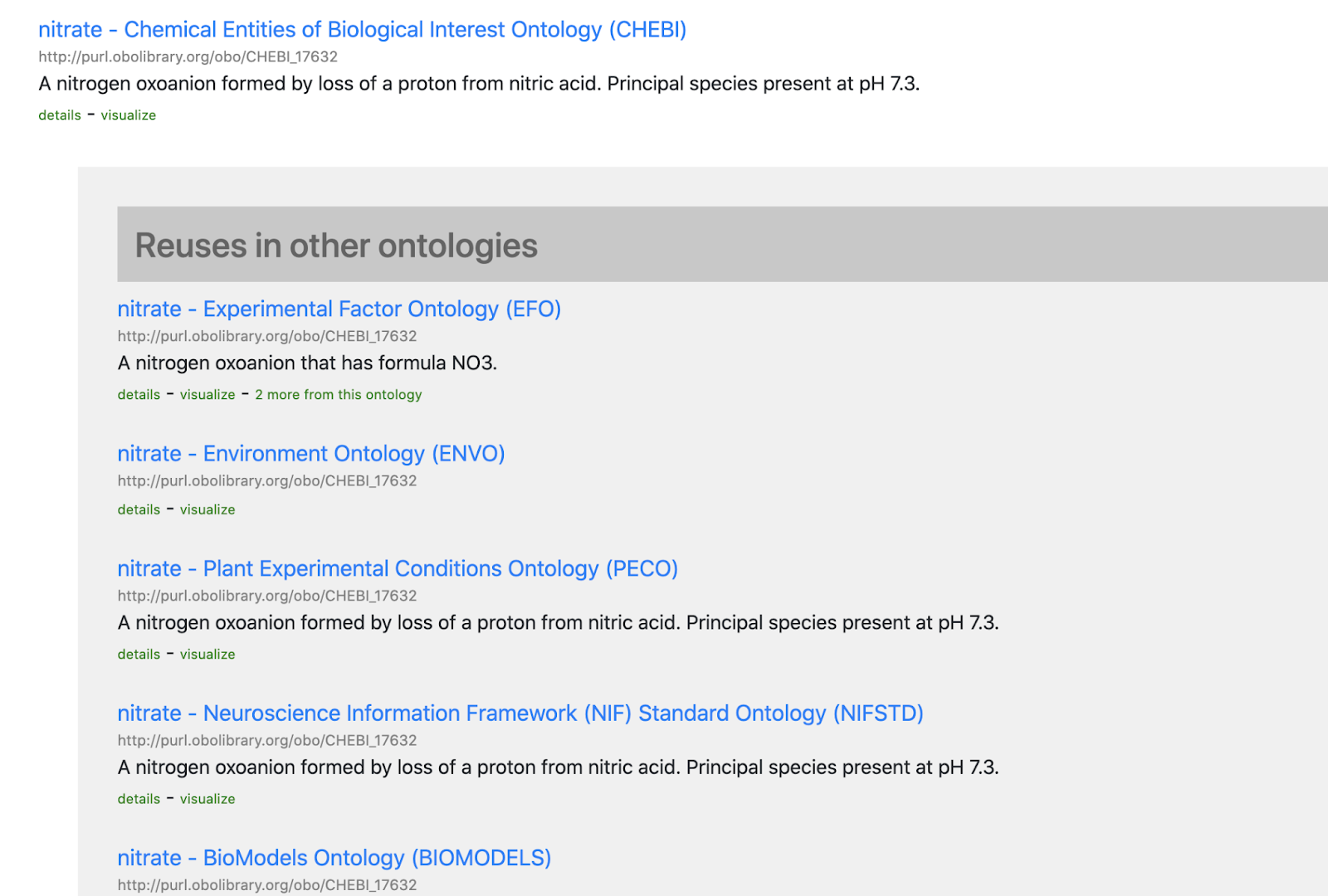
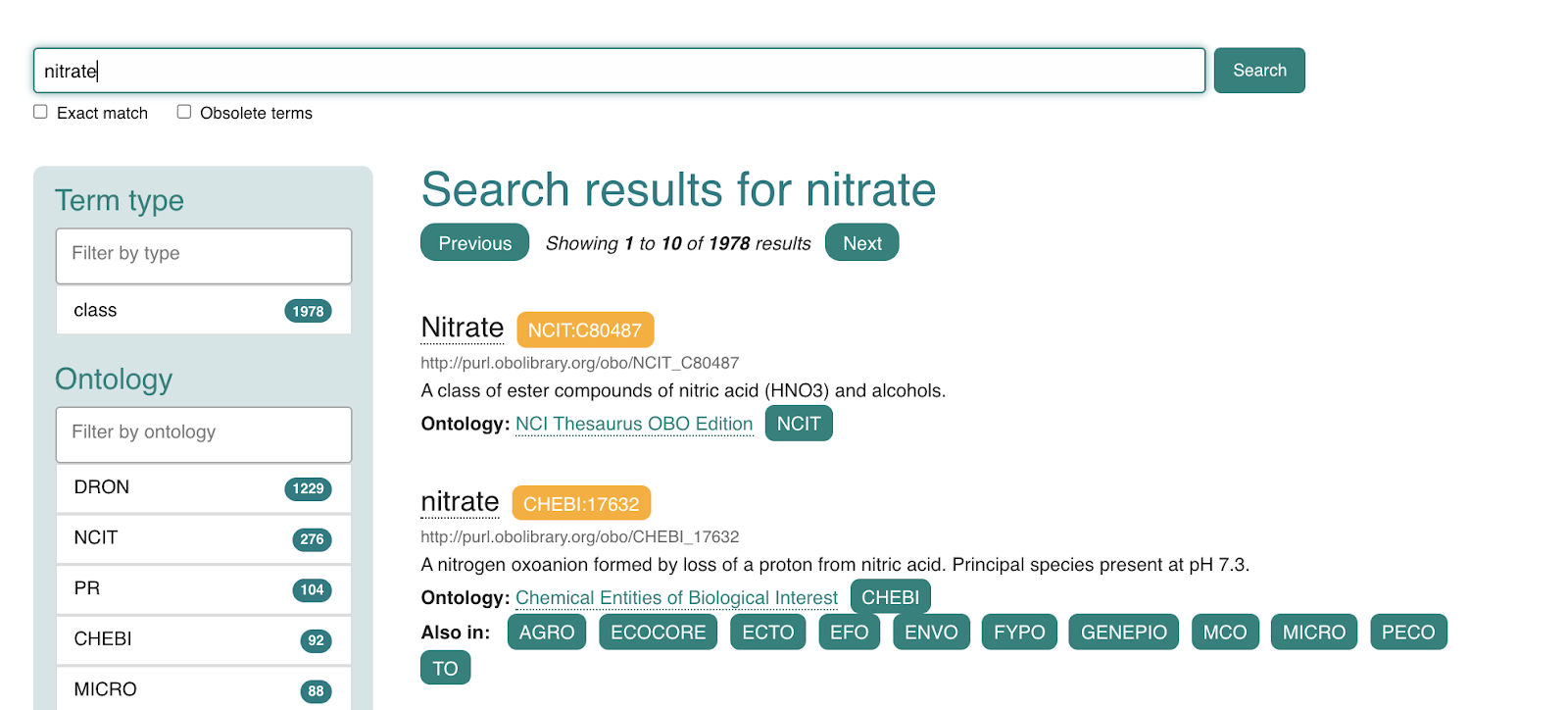
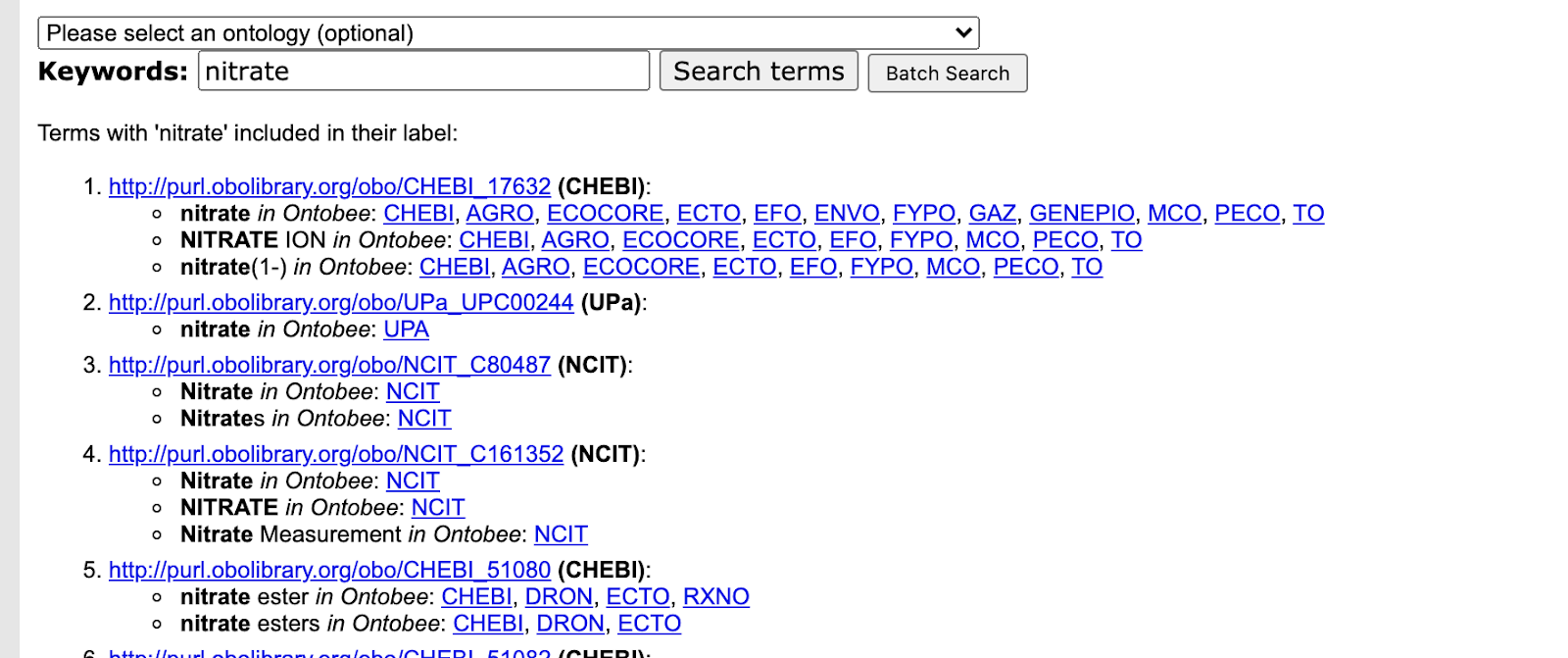
In this post I will describe “Shadow Concepts“, and why they are problematic. In the next forthcoming post I will describe strategies for mitigating these problems, and for avoiding them altogether.
Ontologists love to tease apart the world in fractal ways, considering the many different aspects of concepts and terms we take for granted. This kind of ontological analysis can be extremely helpful in extracting precise statements from ambiguous natural language or fuzzy thinking. For example, when we make statements about a drug like aspirin, do we mean the chemical entity, as represented in CHEBI, or a drug formulation consisting of both active ingredient and filler, as found in DRON? Is aspirin a singular entity, existing in chemical structure space, or is aspirin a set or class whose instances are the quadrillions of actual molecule instances existing in physical-temporal space located inside leaves of willow trees and drug packets?

It can be both illuminating and fun to tease apart both everyday concepts and specialized scientific terms in this way; ‘carving ontology at the joints’ as the ontologists say. Clear thinking and unambiguous communication is undoubtedly important. However, like a lot of ontological analysis, a little moderation goes a long way. This is especially true when it comes to translating this analysis into concrete ontological products intended to be used by non-ontologists. There is a dangerous path that leads to a kind of ontological Balkanization, with concepts fragmented across different disconnected branches of ontologies, or across different ontologies.

This is prevalent in, and as far as I am aware, restricted to the life science / biomedical ontology space (and related environmental/ecological). I am not sure if this is due to something about our domain or knowledge, or more a matter of practice of the groups operating in this space. If this blog post doesn’t describe your experience of ontologies, then it may be less useful for you, although I hope that it will help inoculate you from any future ontologization epidemics.
In this post I will give examples of problems caused by these shadow concepts. But first, I will start by telling an ancient parable.
The Parable of the Intrepid Climber of Mount Ontolympus
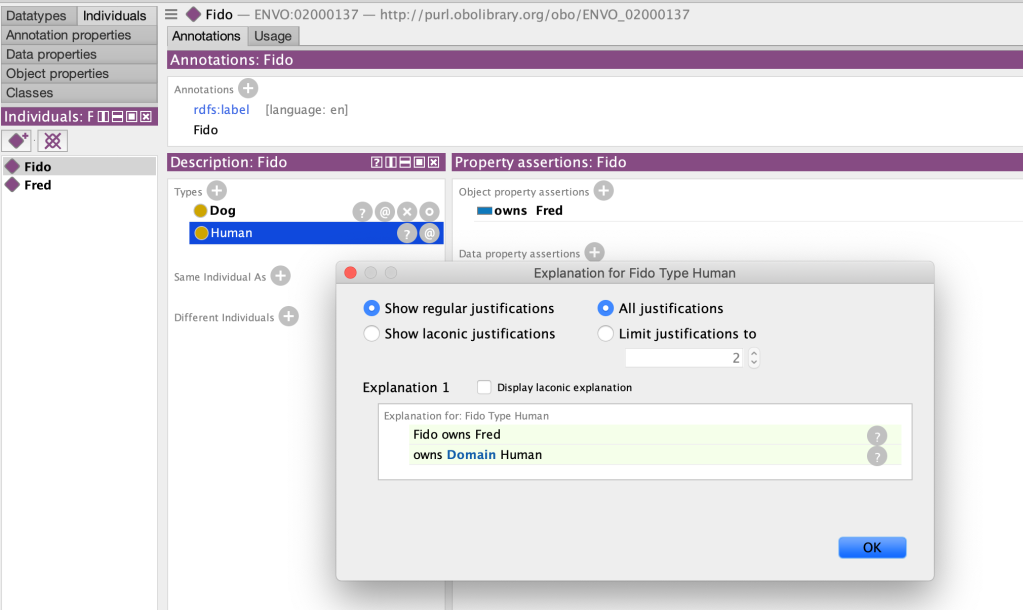
One day an intrepid climber sought to ascend to the peak of the forbidding Mount Ontolympus. However, the climber was ill-prepared, and when the temperature dropped precipitously, he found himself feeling sick, shivering and confused. On performing a temperature assay with a thermometer, he confirmed he was suffering from hypothermia. He cried out for help, comforted in the knowledge that Mount Ontolympus was home to many wandering ontologist-physicians.
On hearing his plaintive cries, an ontologist immediately came to assist. “I have hypothermia, help me” said the climber, knowing it was good to be clear and direct with ontologists. The ontologist smiled and said “you are in luck my friend, for hypothermia is a class in my ontology, where is it clearly a type of measurement. Specifically, a measurement of the temperature of a multicellular organism”. He grabbed the thermometer from the climber and crushed it beneath his feet, exclaiming “without a measurement device, there is no measurement, and thus no hypothermia instance”. The ontologist then triumphantly strode down the mountain, with a shout of “You’re welcome!”.
Seeing the obvious increasing distress of the climber, a second ontologist approached. “I hear you have hypothermia, good sir. You are in luck, for hypothermia is a class in my ontology, where it is clearly a type of information entity, specifically a ‘datum’ that is about a measurement of a temperature of a bodily core. To dispel this instance, you must destroy all hard drives in which the concretizations in which this generically dependent continuant are borne, thus vanquishing the datum”. A troubled look crossed the face of the second ontologists, before he continued: “hmm, neurophysical substrates can also be the bearer of such concretizations,… including the pattern of synaptic wiring in my own brain”. The second ontologist then proceeded to run towards the nearest cliff, which he threw himself from, with the final cry of “your welcome!” echoing throughout the valley below.
The climber was now in peril. A third ontologist approached. She informed the climber: “Sir, you have hypothermia, which is clearly a quality of decreased temperature that inheres in your bodily core. To eliminate this instance, we must increase your body temperature. Here, take this blanket and wrap yourself in it, and sup warm liquids from my flask, …”. But it was too late, the climber’s heart had stopped beating, he had succumbed to his hypothermia, and now lay dead at the third ontologist’s feet.
Upper ontologies lead to shadow concept proliferation
Like all parables, this isn’t intended to be taken literally, and to my knowledge over-ontologization has never led to physical harm beyond moderately increased blood pressure. But there is a core of truth here in how we ontologists are liable to confuse people by insisting on narrowing in on our favorite aspect of a concept, and to waste time chasing different ‘ontological shadows’ rather than focusing on the ‘core’ aspect that matters most to people. This is not to say that it’s not important to distinguish these entities – but there is a time, place, and methodology for doing this.
The Basic Formal Ontology (BFO) is a great tool for analyzing these different aspects of common concepts. It provides a set of upper level categories that subdivide the world roughly similar to Aristotle’s categories. BFO allows us to take something like the class of human hearts and examine the different ontological aspects entailed by the existence of a heart. This includes the site and spatial region at which the heart resides, the disposition of a heart (to pump blood, to make a thumping noise), the realization of those dispositions as processes, the life history of a heart, the representation of a heart such as in a drawing by Vesalius, and so on.
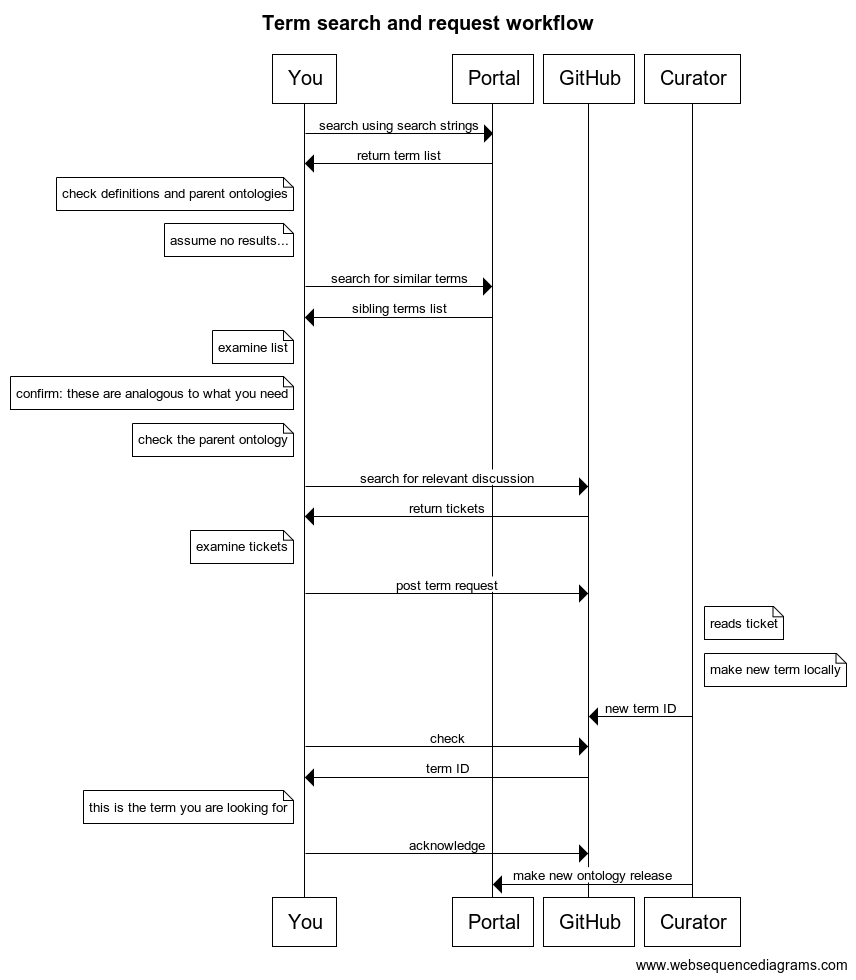
This is illustrated in the following diagram:

BFO is a great tool for analyzing what we mean when we talk about hearts or genes or any other scientific term. Those that are philosophically inclined like to discuss the pros or cons of different upper ontology representations, and there is ongoing vigorous debate about some parts of the above diagram. But that’s not what I am interested in discussing today, and in some ways these debates are irrelevant.
The problem arises when the upper ontology goes beyond an analytic tool, and ontologists start building ontologies that follow this model too literally, creating a vast cross-product of terms that intersect scientific terms such as parts of hearts with shadow ontological aspects. The end result is a ragged lattice, sometimes Balkanized across ontologies, with conceptual drift between the components, confusing users and leading to inconsistency and maintenance tar pits.
Some specific areas where I have seen this problem manifest:
- Geological entities, such as islands (which can be conceived of as material entities in BFO terms), and various shadows such as the area occupied by the island (an immaterial entity).
- Ontologies which follow the OGMS model end up creating many shadow concepts for a disease D, including the disease process, the disease qua disposition, the disease diagnosis (a generically dependent continuant), a concretization of that diagnosis, and many more. For an example of what happens here, try searching CIDO for the different ‘aspects’ of COVID-19
- Biological information macromolecules or macromolecule regions (e.g. DNA regions) can have many aspects, from the material molecular entity itself, through to ‘abstract’ sequences (generically dependent continuants), representation of those sequences in computers (also generically dependent continuants, but concretized by bits and bytes on a hard disk rather than in the mind-independent molecule itself)
- Organismal traits or physical characteristics such as axillary temperature and their information shadows such as axillary temperature measurement datum.
Apologies for the ontological jargon in these descriptions – follow the links to get elucidation of these, but the specific jargon and its meaning isn’t really the point here (although the obfuscated nature of the terminology is a part of the problem).
Shadow concepts lead to conceptual drift and inconsistent hierarchies
Let’s say we have two branches in our ontology, one for material environmental entities such as tropical forests, and another for their immaterial shadows, e.g. tropical forest areas. Ostensibly this is to serve different use cases. An ecologist may be interested in the material entity and its causal influence. A geographer may be interested in the abstracted area as found on a map (I am skeptical about the perceived need for materializing this distinction as separate ontology terms, but I will return to this later).
Problems arise when these branches evolve separately. Let’s say that in response to a request from an ecologist we introduce a subclass ‘tropical broadleaf forest‘ in the material entity hierarchy, but as this didn’t come from a geographer we leave the parallel area hierarchy untouched. Immediately we have a point of confusion – is the omission of a shadow ‘tropical broadleaf forest area‘ class in the area hierarchy intentional, reflecting different use cases between ecologists and geographers, or is it unintentional, reflecting the fact that synchronizing ontology branches is usually poorly handled in ontologies? See The Open World Assumption Considered Harmful for more on this.
The problems worsen if a geographer requests a class ‘broadleaf forest area‘. This seems like it’s broader than our previously introduced ‘tropical broadleaf forest’ class but in fact is broader on a different axis.

This is all very confusing for the typical user who does not care about fine-grained philosophical upper-ontology distinctions and is just looking for terms encompassing ‘tropical forests’. In one hierarchy they get tropical broadleaf forests, in another they don’t get anything relating to broadleaf forests.
This is a small simplified example, the actual confusion caused multiplies when hierarchies grow. Furthermore, other aspects of the hierarchies drift, such as textual definitions, and again it is hard to tell if these differences are intentional reflecting the different ontological nature of the two hierarchies, or if it is just drift.
Needless to say this can cause huge maintenance headaches for ontology developers. This is something we cannot afford, given how few resources and expertise we have for building these ontologies and keeping them up to date.
There are techniques for syncing hierarchies that I will return to later, but these are not cost-free, and have downsides.
Shadow concepts that span ontologies are especially problematic
Syncing shadow concepts within an ontology maintained by a single group is hard enough; problems are vastly exacerbated when the shadows are Balkanized across different ontologies, especially when these ontologies are maintained by different groups.
One example of this is the fragmentation of concepts between OBA (an ontology of biological traits or attributes) and OBI (an ontology biomedical investigations). OBA contains classes that are mostly compositions of some kind of attribute (temperature, height, mass, etc) and some organismal entity or process (e.g. armpit, eye, cell nucleus, apoptosis). OBA is built following the Rector Normalization Pattern using DOSDPs following the EQ pattern.
OBI contains classes representing core entities such as assays and investigations, as well as some datum shadow classes such as axillary temperature measurement datum, defined as A temperature measurement datum that is about an axilla.
Immediately we see here there is a problem from the perspective of ontology modularization. Modularization is already hard, especially when coordinated among many different ontology building groups with different objectives. But ostensibly in OBO we should have ‘orthogonal’ ontologies with clear scope. And on the one hand, we do have modularization and scope here: datums (as in datum classes, not actual data) arising from experimental investigations go in OBI, the core traits which those data are about go in OBA.
But from a maintenance and usability perspective this is really problematic. It is especially hard to synchronize the hierarchies and leverage work done by one group for another due to the fact the terms are in different ontologies with different ontology building processes and design perspectives. Users are frequently confused. Many users take a “pick and mix” approach to finding terms, ignoring ontological fine details and collecting terms that are ‘close enough’ (see How to select and request terms from ontologies). Rather than request a term ‘axillary temperature’ from OBA they may take the OBI term, and end up with a mixture of datum shadows and core concepts. This actually has multiple negative ramifications from multiple perspectives: reasoning, text mining, search, usability. It also goes against plain common sense – things that are alike should go in the same ontology, things that are unalike go in different ones.
Case Study: The Molecular Sequence Ontology
A classic shadow concept use case is distinguishing between the molecular aspect of genomic entities (genes, gene products, regulatory regions, introns, etc) and their sequence/information aspect. This is not idle philosophizing – it lies at the heart of eternal questions of ‘what is a gene‘. It is important for giving clear answers to questions like ‘how many genes in the human genome’.
I won’t go into the nuances here, interesting as they are. The important thing to know here was that the Sequence Ontology (SO) was originally formally ontologically uncommitted with regards to BFO. What this means is that neither the genomics use case driven developers of SO not the users particularly cared about the distinction between molecules and information. One of the main uses for SO is typing features in GFF files, querying genomics databases, and viewing sequences in genome browsers (this probably makes it the most widely instantiated biological ontology, with trillions of SO instances, compared with the billions in GO). Users are typically OK with a gene simultaneously being an abstract piece of information carried by DNA molecules, an actual molecular region, and a representation in a computer. But this led to a few complications when working with an ontology.
To rectify this, in 2011, I proposed the creation of a ‘molecular sequence ontology‘. I am quite proud of the paper and the work my colleagues and I did on this, but I take full responsibility for the part of this paper that proposed the ‘SOM’ (Sequence Ontology of Molecules, later MSO), which I now regard as a mistake. The basic idea is captured in Table 2 of the paper.

the basic idea was to ‘split’ SO into two ontologies that would largely shadow each other, one branch encompassing information entities (the SO would be ‘recast’ as this branch), and another branch for the molecular entities (MSO).
A number of good papers came out of this effort:
Intelligently Designed Ontology Alignment: A Case Study from the Sequence Ontology
Michael Sinclair, Michael Bada, and Karen Eilbeck
ISMB Bio-ontologies track conference proceeding 2018
Efforts toward a More Consistent and Interoperable Sequence Ontology
Michael Bada and Karen Eilbeck. Conference Proceeding ICBO 2012
Toward a Richer Representation of Sequence Variation in the Sequence Ontology
Michael Bada and Karen Eilbeck. Annotation, Interpretation and Management of Mutations. A workshop at ECCB10.
These are definitely worth reading for understanding the motivation for MSO, the challenges for developing and synchronizing it, and improvements that were made. However, ultimately the challenges in syncing the ontologies proved greater than expected, and did not stack well against the benefits, and resources ran out before a number of the benefits could be realized.
At the Biocuration workshop in 2017 there was a birds of a feather session featuring the MSO developers plus a large cross-representation of the curators that use SO for annotating genomic data. The outcome was that not everyone saw the need for splitting into two ontologies, and many found the ontological motivation confusing. However, everyone saw the value of the ontology editing effort that had gone into MSO (which included many improvements orthogonal to the ontological recasting), and so the consensus was to fold in any changes made into MSO back into SO that did not pertain to the fine-grained MSO/SO distinction. Unfortunately this was quite challenging as the ontologies had diverged in different ways, and not all the changes have been folded back in.
I think there is a lesson there for us all — we should make sure we have buy-in from actual users and not just ontologists before making decisions about how to shape and recast our ontologies. “Ontological perfection” can seem a lofty goal, especially for newcomers to ontology development, but this has to be balanced against practical use cases. And crucially we should be careful not to underestimate development, maintenance and synchronization costs. It’s easy to ignore these especially when building something from scratch, but just like in software engineering, maintenance eats up the lions share of the cost of developing an ontology.
Strategies for managing and avoiding shadow concepts
In the next post I will provide some strategies for mitigating and avoiding these problems.
Where shadow concepts are unavoidable, templating systems like DOSDPs should be used to sync the hierarchies (ideally all aspects of the shadow, including lexical information, should mirror the core term). This is necessary but not sufficient. There needs to be extra rigor and various workflow and social coordination processes in place to ensure synchrony, especially for inter-ontology shadows. There are a lot of reasoning traps the unwary can fall into.
I will also review various conflation techniques including what I call the Schulzian transform. These attempt to “stitch together” fragmented classes back into intuitive concepts.
I will also review cases where I think materialization of certain classes of shadow are genuinely useful. These include things such as genes and their ‘reference’ proteins, genes and their functions, or infectious diseases like COVID-19 and the taxonomic classification of their agents like SARS-CoV-2. Nevertheless it can still be useful to have strategies to selectively conflate these.
But really these mitigation and conflation techniques should not be necessary most of the time: in most cases there is no need to create shadow hierarchies. I can’t emphasize this strongly enough. If you want to talk about a COVID diagnosis (which is undoubtedly a different ontological entity than the instance of the disease itself) then you don’t need to materialize a shadow of the whole disease hierarchy. Just use the core class! You can represent the aspect separately as ‘post-composition’. The same goes if you feel yourself wanting classes with the string “datum” appended at the end. Instead of making a semi-competing shadow ontology, work on the core ontology, and if you really really need to talk about the “X datum” rather than “X” itself (and I doubt you do), just do this via post-coordination. I will show examples in the next post, but in many ways this should just be common sense.